bioinformatics
Single-sample GSEA is now in Orange
Martin Špendl
Feb 23, 2023
In the world of biology, everything revolves around genes. They carry information about the essential functions of cells and create mRNA molecules that execute these functions. Biologists like to extract information about gene expression by simply counting the number of mRNA molecules produced by genes. There are more than 20,000 genes in the human genome, which makes the expression matrix very wide. Biologists like identifying important genes in different processes and grouping them into gene sets. By comparing their expression over different samples, they can gain insight into the mechanism of change.
One of the widely used methods for assigning a value to the expression of a gene set is the single-sample extension of Gene Set Enrichment Analysis (ssGSEA), proposed by Barbie et al.. The ssGSEA method gives a score related to the overexpression of genes in an individual sample, contrary to GSEA, which calculates the change across multiple samples. The algorithm sums the contributions of genes related to their rank in an ordered expression matrix. High expression values contribute positively to the score, while lower values contribute negatively.
The method was first introduced as a signature projection method and quickly gained popularity for assigning gene set scores to samples. It’s most widely used in cancer biology to discover novel tumour subtypes, search for new prognostic markers and uncover the underlying tumour-driving process. It’s now available in Orange as a Single sample scoring widget from the Bioinformatics add-on.
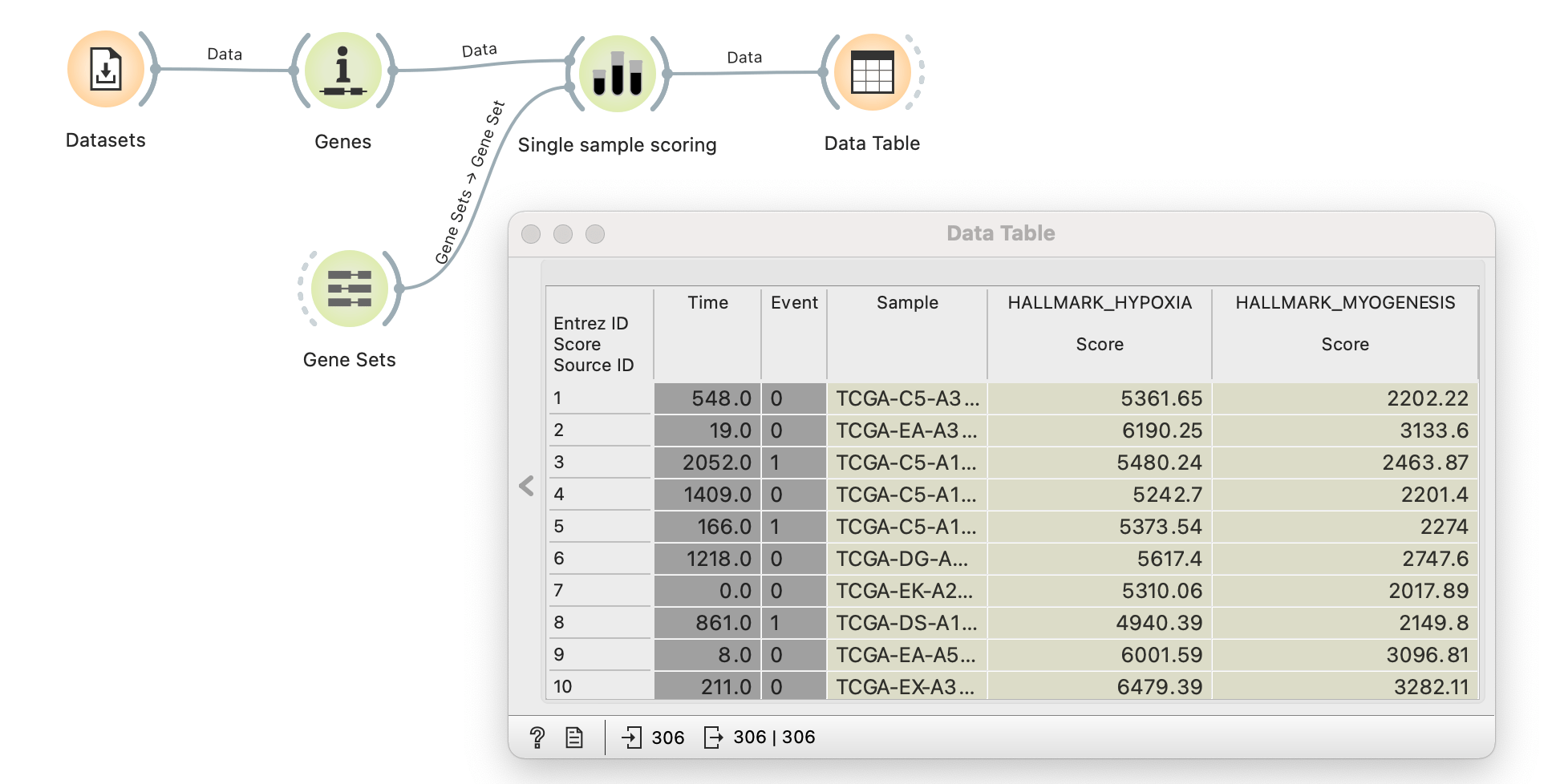
The illustration above demonstrates how to perform single-sample GSEA analysis on the TCGA Cervical carcinoma data (TCGA-CESC) from the Datasets widget. ssGSEA method is the default option for scoring in the Single sample scoring widget, which takes two parameters: expression data through the Genes widget and gene sets through the Gene Sets widget. As seen in the Data Table widget, sample scores are added as metadata parameters to the original data. Users can now easily compute gene set scores and enrich their analysis.